
 Ubuntu кластер
Ubuntu кластер
 *** ЧТО ТАКОЕ UBUNTU?
*** ЧТО ТАКОЕ UBUNTU?
Ubuntu - это разрабатываемая сообществом, основанная на ядре Linux операционная система, которая идеально подходит для использования на персональных компьютерах, ноутбуках и серверах. Она содержит все необходимые программы, которые вам нужны: программу просмотра Интернет, офисный пакет для работы с текстами, электронными таблицами и презентациями, программы для общения в Интернет и много других.
Дистрибутивы семейства Ubuntu являются комплектом специально подобранного ПО. Это операционная система плюс множество нужных и полезных программ, которые свободно и без ограничений могут быть использованы вами и вашими друзьями и коллегами. Базовая полнофункциональная версия системы для персональных компьютеров и ноутбуков или версия для серверов занимают всего 1 CD, поэтому вы легко можете получить её для себя и делиться с окружающими. Специальные DVD версии дистрибутива включают практически всё ПО, которое вам может понадобиться для решения любых повседневных задач.
*** ПОДГОТОВКА КЛАСТЕРА
Для установки линукс-кластера на основе дистрибутива Ubuntu вам потребуется установочный диск Ubuntu Desktop Edition, с помощью которого вы установить операционную систему на консоль (главный компьютер) кластера. С того же самого диска нужно будет установить операционную систему на вычислительные узлы кластера. Однако, если вычислительные узлы не предполагается использовать в качестве офисных машин, но только как узлы кластера, то на эти компьютеры правильнее будет установить серверную редакцию дистрибутива - Ubuntu Server Edition, не содержащую графического интерфейса, и соответственно более легкую и быструю.
Сеть кластера следует спроектировать так, чтобы все узлы имели доступ в интернет. Это нужно для большего комфорта при настройке кластера и установки необходимого программного обеспечения на его узлах. Дело в том, что установка ПО в Ubuntu выполняется посредством закачки новейших версий необходимых пакетов из внешних репозиториев. В действительности достаточно будет обеспечить выходом в Интернет только главный компьютер (консоль кластера), а для вычислительных узлов необходима только возможность загружать программные пакеты через прокси. Осуществить это можно, установив на консоли кластера пакет apt-cacher-ng, а на вычислительных узлах сконфигурировать менеджер пакетов apt-get для работы через прокси. Для этого на консоли кластера выполним команду:
sudo apt-get install apt-cacher-ng
После этого на всех остальных узлах выполним команды:
sudo sh -c "echo \"Acquire::http { Proxy \\\"http://192.168.1.1:3142\\\"; }; \"> /etc/apt/apt.conf.d/01proxy"
sudo apt-get update
Здесь мы предполагаем, что адрес 192.168.1.1 - это адрес консоли кластера во внутренней сети кластера.
После установки базовой операционной системы необходимо доустановить необходимое для функциониования кластера по: компиляторы, библиотеки и программы OpenMPI, SSH, NFS. Выполнить это можно с помощью следующих команд:
sudo apt-get install mc build-essential fort77 gfortran libstdc++5 libltdl7-dev
sudo apt-get install openssh-server nfs-kernel-server
sudo apt-get install openmpi-bin openmpi-doc
Данная последовательность команд устанавливает в систему OpenMPI и компиляторы из набора Gnu Compiller Collection (gcc). Компиляторы gcc обладают одним существенным (в некоторых случаях) недостатком. В них нет поддержки работы с типами данных REAL*16 и COMPLEX*32 (в терминологии Фортрана). Если для ваших задач необходима такая точность вычислений, то вы вместо стандартного набора компиляторов и пакета OpenMPI из состава дистрибутива должны будете установить компиляторы фирмы Intel и скомпилировать OpenMPI с поддержкой этих компиляторов. Как это сделать - рассказано в следующих двух параграфах этой статьи. Если же точность REAL*8 и COMPLEX*16 вас устраивает, то следующие два параграфа вы можете пропустить.
ВАЖНО! Если вы решили использовать компиляторы фирмы Intel, то не следует устанавливать OpenMPI из состава дистрибутива. То есть команду sudo apt-get install openmpi-bin openmpi-doc (см. выше) выполнять не следует.
*** УСТАНОВКА КОМПИЛЯТОРА INTEL FORTRAN
Скачать свежую версию компилятора можно с официального сайта компании Intel, с этой страницы. Получив и разархивировав в своем домашнем каталоге архив с компилятором, выполним процедуру установки, запустив скрипт install.sh, который находится в каталоге, получившимся после разархивации. При запуске скрипт спросит вас о том, в каком варианте произвести установку компилятора. Нужно выбрать вариант N3 - установку в пользовательский каталог. Для ответа на все дальнейшие вопросы программы следует выбирать вариант, предложенный программой по умолчанию.
После окончания установки, чтобы все заработало необходимо поправить некоторые переменные окружения. Для этого исправим файл .bashrc, находящийся в домашнем каталоге пользователя, используя следующую команду (не забыв после передодключиться в систему):
echo ". $HOME/intel/Compiler/11.1/046/bin/ifortvars.sh ia32" >> $HOME/.bashrc
Эта команда построена в предоложении, что используется версия 11.1.046 компилятора. Если же к тому моменту, когда вы воспользуетесь данными рекомендациями, фирма Intel выпустит новую версию, то команду надо будет поправить соответствующим образом.
*** УСТАНОВКА OpenMPI ДЛЯ КОМПИЛЯТОРА INTEL
К сожалению пакет OpenMPI, имеющийся в дистрибутиве Ubuntu, не знает о существовании компилятора Intel. Поэтому его придется установить из исходников, которые можно загрузить с официального сайта проекта OpenMPI. Разархивировав загруженный файл, перейдем в полученный в результате этого каталог исходников и выполним процедуру установки, последовательно запустив на исполнение следующие команды:
./configure --prefix=$HOME/openmpi F77=ifort FC=ifort
make
make install
В случае, если вы намереваетесь использовать и установили кроме компилятора Intel Fortran, так же и Intel С++, то команда конфигурации должна будет выглядеть так:
./configure --prefix=$HOME/openmpi CC=icc CXX=icpc F77=ifort FC=ifort
По окончании процесса установки, как и в случае с компилятором, необходимо поправить некоторые переменные окружения. Делаем это с помощью следующих команд модифицируем файл .bashrc:
echo "PATH=$HOME/openmpi/bin:\$PATH" >> $HOME/.bashrc
echo "export PATH" >> $HOME/.bashrc
echo "LD_LIBRARY_PATH=$HOME/openmpi/lib:\$LD_LIBRARY_PATH" >> $HOME/.bashrc
echo "export LD_LIBRARY_PATH" >> $HOME/.bashrc
По окончании процедуры необходимо переподключиться в систему (перелогиниться).
*** НАСТРОЙКА СЕТИ КЛАСТЕРА
Для того, чтобы MPI имела возможность без дополнительных запросов запускать части параллельной прогаммы на вычислительных узлах кластера, вы должны будете организовать беспарольный доступ по протоколу SSH с консоли кластера на все вычислительные узлы. Как это сделать - читайте соответствующий раздел этого сайта.
Следующим нашим шагом будет организация общего для всех узлов кластера каталога, где будут храниться исходники параллельных программ, их исполняемые модули и файлы с данными. Сделать это можно, настроив сетевую файловую систему NFS. Для простоты общий каталог сделаем подкаталгом домашне директории пользователя. Назовем его к прмеру "mpi".
Предположим, что узлы кластера имеют адреса внутри сети 192.168.1.0/255.255.255.0, и консоль кластера имеет адрес 192.168.1.1.
На консоли кластера, выполнив следующую команду, добавляем строчку в файл /etc/exports для создания сетевого ресурса:
sudo sh -c "echo \"$HOME/mpi 192.168.1.0/24(rw,insecure,nohide,all_squash,anonuid=1000,anongid=1000,no_subtree_check)\" >> /etc/exports"
После этого необходимо перезапустить сервис NFS:
sudo /etc/init.d/nfs-kernel-server restart
На всех остальных узлах кластера мы должны будем поправить файл /etc/fstab, добавив туда запись для автоматического монтирования сетевого ресурса. Делается это командой:
sudo sh -c "echo \"192.168.1.1:$HOME/mpi $HOME/mpi nfs rsize=8192,wsize=8192,rw,auto,exec 0 0\" >> /etc/fstab"
Теперь для проверки перезагрузим вычислительные узлы кластера. После перезагрузки каталог mpi в домашней директории пользователя должен быть смонтирован и его содержимое должно быть эквивалентно содержимому такого же каталога на консоли кластера.
Если вы решили использовать компилятор Intel Fortran/Intel C++ и, соответственно, на консоли кластера установили этот компилятор и OpenMPI, скомпиленный из исходников, то то же самое вы должны сделать на всех вычислительных узлах. Однако последнее - не обязательно. Вместо этого вы можете подключить по сети (через NFS) каталоги с установленными компилятором и OpenMPI (openmpi и intel) аналогично тому, как вы только что это сделали подключая рабочий каталог mpi. В дополнение к этому вам необходимо подкорректировать на всех вычислительных узлах кластера файл /etc/environment следующими командами:
sudo sed -i -e 's;PATH=";PATH="'"$HOME"'/openmpi/bin:'"$HOME"'/intel/Compiler/11.1/046/bin/ia32:;' /etc/environment
sudo sh -c "echo \"LD_LIBRARY_PATH=\\\"$HOME/openmpi/lib:$HOME/intel/Compiler/11.1/046/lib/ia32:$HOME/intel/Compiler/11.1/046/lib/32\\\"\" >> /etc/environment"
*** НАСТРОЙКА ПАРАЛЛЕЛЬНОЙ СРЕДЫ
Настройка OpenMPI максимально проста. Вы должны в любом удобно месте создать текстовый файл (назовем его например mpi.hosts), в который запишете список всех узлов вашего кластера, как вычислительных узлов, так и консоли. Для простоты будем считать, что вы перечислите там ip-адреса узлов. Один узел - одна строчка. В самом простом варианте конфигурации кластера этого будет достаточно для начала работы.
*** КОМПИЛЯЦИЯ ПАРАЛЛЕЛЬНОЙ ПРОГРАММЫ
Для примера и для проверки работоспособности кластера скачаем в каталог mpi тестовую программу
вычисления числа 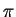 flops.f.
Для компиляции необходимо выполнить следующую простую команду:
flops.f.
Для компиляции необходимо выполнить следующую простую команду:
mpif77 flops.f -o flops
В результате должен получится исполняемый файл с именем "flops". Для запуска его в локальном однопроцессорном режиме достаточно запустить этот файл на исполнение обычным образом, то есть выполнить команду ./flops. На выходе мы получим информацию о скорости работы программы. Если все прошло нормально, то можем переходить к тестированию кластера.
*** ЗАПУСК ПАРАЛЛЕЛЬНОЙ ПРОГРАММЫ
Для запуска нашей программы параллельно на нескольких процессорах (узлах кластера) необходимо использовать диспетчер задач MPI или менеджер очередей, например Torque. Про менеджер очередей и ресурсов Torque мы поговоирм позже. Пока же обойдемся без него. Нам понадобится созданный нами ранее файл-список узлов mpi.hosts. С помощью этого списка запуск нашей параллеььной программы осуществляется командой:
mpiexec -hostfile mpi.hosts -np 5 ./flops
Параметр -np 5 означает, что программа будет запущена на пяти узлах кластера. Отработав, программа выдаст на экран информацию о скоростных характеристаках кластера в целом и отдельных его узлов.
В случае, если ваша параллельная программа состоит из отдельных программных модулей, то имеется возможность запускать из вместе, как единый комплекс. Предположим, что ваша программа состоит из двух модулей - visio и calc. Модуль calc - это собственно параллельная программа, которая в многопроцессороном режиме обрабатывает большой массив данных, и которая занимает 5 процессоров кластера. Модуль visio - программа визуализации полученных данных. Программа visio должна работать на одном процессоре. Запуск такой комплексной программы будет выглядеть так:
mpiexec -hostfile mpi.hosts -np 1 ./visio : -np 5 ./calc